

Dr. Kristin Forssmann, direttore sviluppo clinico Meditrial.
Dopo oltre 20 anni, è entrata in vigore in Europa una nuova legislazione, il “regolamento sui dispositivi medici” (EU‐MDR 2017/745). I fattori scatenanti l’avvio e l’attuazione di questa legge europea, molto più severa rispetto alle precedenti direttive, sono stati gli scandali relativi alla sicurezza dei dispositivi medici, ad esempio le protesi mammarie difettose realizzate con materiali scadenti, o le protesi dell’anca ritirate dal mercato poiché richiedevano un secondo intervento in un numero molto elevato di pazienti.
Il regolamento MDR è il quadro normativo che definisce legalmente la produzione e la distribuzione dei dispositivi medici in Europa. La conformità a questa normativa è obbligatoria per le aziende produttrici di dispositivi medici (fabbricanti legali) in commercio negli Stati Membri dell’Unione Europea. Il nuovo Regolamento richiede da un lato, che le autorità di vigilanza collaborino in modo armonizzato e, dall’altro, che i fabbricanti mettano in campo strategie per monitorare l’efficacia e la sicurezza dei dispositivi medici dopo la commercializzazione, in modo più efficiente e rigoroso.
Relazione tra la sorveglianza e il piano di follow-up clinico
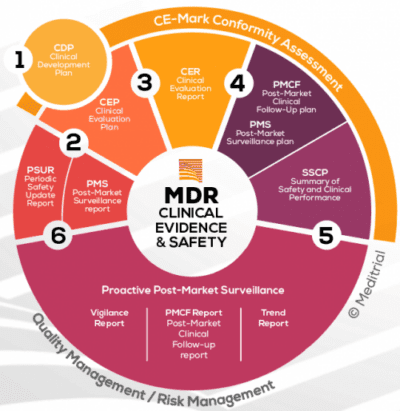
La nuova situazione giuridica introdotta dal regolamento MDR mira a consentire agli organismi notificati e alle autorità di sorveglianza di svolgere correttamente i propri compiti. In questo contesto, la sorveglianza post‐commercializzazione (PMS) svolge un ruolo molto importante come elemento centrale, per la cui attuazione, a seconda della classe di rischio, è necessario condurre i cosiddetti studi di follow‐up clinico post‐commercializzazione (PMCF) al fine di monitorare costantemente l’efficacia clinica e la sicurezza dei dispositivi medici interessati.
La nuova legislazione ha perfezionato e ampliato la necessità per i produttori di disporre di un sistema di sorveglianza post-commercializzazione (PMS). Secondo questa normativa, è richiesto anche un piano di follow-up clinico post-commercializzazione (PMCF). I produttori di dispositivi medici ad alto rischio sono obbligati sia a condurre il follow-up clinico post-commercializzazione, sia di raccogliere e analizzare dati di sorveglianza o PMS. Vi è quindi la necessità di generare evidenze dai dati clinici

Registri modulari: una soluzione intelligente ed efficiente
Per i fabbricanti di dispositivi medici, la necessità di condurre diversi studi per la sorveglianza e il follow-up clinico post-commercializzazione, è notevolmente impegnativa e costosa. Un recente articolo ha proposto una soluzione intelligente per gestire al meglio l’impegno della generazione di evidenze.
L’articolo “A Modular Approach to Combine Postmarket Clinical Follow‐Up Studies and Postmarket Surveillance Studies” 1 propone un approccio che i fabbricanti possono attuare, per assolvere i loro obblighi di sorveglianza del mercato non in singoli studi PMCF, ma sotto forma di registri strutturati in modo modulare (vedi figura). Il concetto modulare combina la sorveglianza post‐commercializzazione ed il follow‐up clinico post‐commercializzazione. Ad esempio, si parte dall’elaborazione dei parametri fondamentali per la sicurezza (SC 1‐SC 3) e altrettanti parametri relativi alla funzione del dispositivo (Fct 1‐Fct 3). Nell’esempio illustrato in figura, vengono valutate due popolazioni (adulti e bambini). Lo studio viene condotto in due paesi, ciascuno con 4 centri.
A causa delle diverse normative dei due paesi, e della specializzazione dei centri (che possono trattare solo adulti, solo adolescenti, o entrambi), talvolta gli sponsor organizzano multipli studi. Questa frammentazione è costosa e può creare difficoltà nell’analisi dei dati e nell’accettazione delle evidenze da parte degli organismi di certificazione. L’approccio modulare, invece, consente di standardizzare la raccolta delle evidenze, applicando un set unico di quesiti di sicurezza o funzionalità in varie combinazioni appropriate, ma utilizzando uno stesso protocollo clinico.
Approccio modulare per la sorveglianza e gli studi di follow-up post-commercializzazione

Il protocollo “universale” post-commercializzazione
Gli autori dello studio sopracitato hanno sviluppato un modello del protocollo clinico (clinical investigation plan o CIP) che rendono pubblicamente disponibile come appendice alla loro pubblicazione. Il modello CIP è concepito come base per un approccio modulare che congiunge la sorveglianza (PMS) e il follow-up clinico (PMCF) sotto un unico ombrello.
Tale modello è sviluppato in conformità con il regolamento MDR, in particolare l’Allegato XV, Sezione 3.8. Esso è anche in linea con la norma EN ISO 14155 (Allegato A) che fornisce il quadro di riferimento per la pianificazione, conduzione e rendicontazione dei dispositivi medici. Nello sviluppo del modello, sono stati integrati anche i punti relativi a SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) e SPIRIT-PRO, con una lista di 33 elementi raccomandati. Di recente, la dichiarazione SPIRIT è stata ampliata con riferimenti all’inclusione degli esiti riferiti dai pazienti (PRO), denominati SPIRIT-PRO. Poiché nessuna di queste linee guida fornisce uno schema di un protocollo di studio, il template di protocollo può essere utile agli sponsor per strutturare correttamente gli studi interventistici e osservazionali.
Implementazione nella pratica clinica per i dispositivi medici
Un esempio di applicazione dell’approccio modulare è stato messo in opera in un registro osservazionale di sorveglianza post‐commercializzazione che include soggetti trattati con impianti ortopedici biodegradabili a base di magnesio di classe III specifici per l’uso previsto. L’obiettivo di questo registro clinico è quello di raccogliere sistematicamente dati osservazionali sulla sicurezza, la qualità del prodotto, la funzione intenzionale e la qualità generale della vita derivanti dall’uso di routine degli impianti ortopedici in più centri. Il registro prevede la raccolta dati dal momento precedente l’intervento fino a 5 anni dopo l’intervento.
Questo modello di registro è stato applicato con successo anche in registri di sorveglianza dei farmaci e rappresenta un valore aggiunto sia per i pazienti che sono facilitati nel partecipare, sia per le aziende che desiderano una estensione delle indicazioni, ad esempio per le malattie rare.
Come creare un programma clinico sostenibile

Un’attenzione particolare nei registri è rivolta al paziente2, perché la prima priorità è dimostrare i benefici con un diretto impatto sulla salute e sulla qualità di vita. Ciò può essere fatto con l’aiuto di speciali questionari sulla qualità della vita e sui sintomi specifici della malattia, che devono essere convalidati in ogni singola lingua. Il valore e l’importanza di tali strumenti sono enormi se possono fornire dati affidabili e riproducibili.
Gli studi post-commercializzazione di grandi dimensioni richiedono soluzioni moderne e complete per la raccolta dei dati clinici. Tecnicamente, i registri possono essere mappati in modo eccellente nei sistemi di “electronic data capture” (EDC). Gli strumenti digitali oggi offrono la possibilità di semplificare l’acquisizione di dati clinici e immagini diagnostiche, il monitoraggio remoto, l’analisi dei dati automatica e la valutazione della sicurezza. Inoltre, sono ora disponibili App da scaricare sul proprio cellulare, in cui il paziente può dare il proprio consenso e non deve nemmeno recarsi al centro di analisi per raccogliere i questionari. Naturalmente, le leggi e le normative sulla protezione dei dati devono essere rigorosamente rispettate, il tutto per la sicurezza nell’interesse del paziente.
Autore: Dr. Kristin Forßmann, direttore sviluppo clinico Meditrial GmbH, Berlino, Germania
Template del protocollo modulare (Indice dei contenuti). Ziegler et al, 2021






Leave A Comment