
After more than 20 years, new legislation, the “Medical Device Regulation” (EU-MDR 2017/745), has come into force in Europe. The triggers for the initiation and implementation of this European law, which is much stricter than previous directives, were scandals related to the safety of medical devices, e.g., defective breast implants made from substandard materials, or hip implants withdrawn from the market because they required a second surgery in a very large number of patients.
The MDR regulation is the regulatory framework that legally defines the production and distribution of medical devices in Europe. Compliance with this regulation is mandatory for medical device companies (legal manufacturers) on the market in EU member states. The new Regulation requires, on the one hand, that supervisory authorities cooperate in a harmonized manner and, on the other hand, that manufacturers put in place strategies to monitor the effectiveness and safety of medical devices after marketing in a more efficient and rigorous manner.
RELATIONSHIP BETWEEN SURVEILLANCE AND CLINICAL FOLLOW-UP PLAN
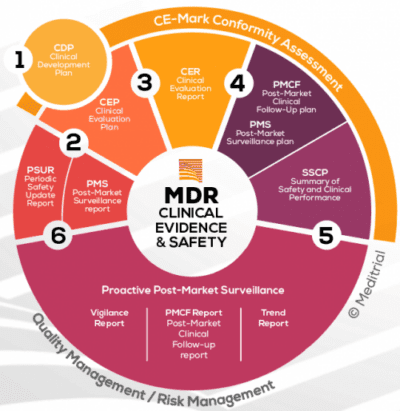
The new legal situation introduced by the MDR Regulation aims to enable notified bodies and surveillance authorities to properly carry out their tasks. In this context, post-market surveillance (PMS) plays a very important role as a central element, for the implementation of which, depending on the risk class, it is necessary to conduct so-called post-market clinical follow-up studies (PMCFs) in order to continuously monitor the clinical efficacy and safety of the medical devices concerned.
The new legislation has refined and expanded the need for manufacturers to have a post-marketing surveillance system (PMS). Under this legislation, a post-marketing clinical follow-up plan (PMCF) is also required. Manufacturers of high-risk medical devices are required both to conduct post-marketing clinical follow-up and to collect and analyze surveillance data or PMS. There is therefore a need to generate evidence from clinical data

MODULAR REGISTERS: AN INTELLIGENT AND EFFICIENT SOLUTION
For medical device manufacturers, the need to conduct multiple studies for post-marketing clinical surveillance and follow-up is significantly challenging and costly. A recent article proposed a smart solution to better manage the evidence generation effort.
The article “A Modular Approach to Combine Post-Market Clinical Follow-Up Studies and Post-Market Surveillance Studies” 1 proposes an approach that manufacturers can implement, to fulfill their market surveillance obligations not in individual PMCF studies, but in the form of modularly structured registries (see figure). The modular concept combines post-marketing surveillance and post-marketing clinical follow-up. For example, it starts with the development of key safety parameters (SC 1-SC 3) and as many parameters related to device function (Fct 1-Fct 3). In the example shown in the figure, two populations (adults and children) are evaluated. The study is conducted in two countries, each with 4 centers.
Because of the different regulations in the two countries, and the specialization of the centers (which may treat only adults, only adolescents, or both), sometimes sponsors organize multiple studies. This fragmentation is costly and can create difficulties in data analysis and acceptance of evidence by certifying bodies. The modular approach, on the other hand, allows standardization of evidence collection by applying a single set of safety or functionality questions in various appropriate combinations, but using the same clinical protocol.
Modular approach for surveillance and post-marketing follow-up studies

THE POST MARKETING “UNIVERSAL” PROTOCOL
The authors of the above study have developed a clinical protocol model (clinical investigation plan or CIP) that they make publicly available as an appendix to their publication. The CIP model is designed as the basis for a modular approach that combines surveillance (PMS) and clinical follow-up (PMCF) under one umbrella.
This model is developed in accordance with the MDR regulation, in particular Annex XV, Section 3.8. It is also in line with EN ISO 14155 (Annex A), which provides the framework for planning, conducting and reporting on medical devices. In developing the model, SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) and SPIRIT-PRO were also incorporated, with a list of 33 recommended items. Recently, the SPIRIT statement was expanded with references to the inclusion of patient-reported outcomes (PROs), referred to as SPIRIT-PRO. Since none of these guidelines provide an outline of a study protocol, the protocol template may be useful for sponsors to properly structure interventional and observational studies.
IMPLEMENTATION IN CLINICAL PRACTICE FOR MEDICAL DEVICES
An example of the application of the modular approach was implemented in an observational post-market surveillance registry that includes subjects treated with biodegradable magnesium-based class III orthopedic implants specific to their intended use. The goal of this clinical registry is to systematically collect observational data on safety, product quality, intended function, and overall quality of life from routine use of orthopedic implants at multiple centers. The registry involves data collection from the time before surgery to 5 years after surgery.
This registry model has also been successfully applied in drug surveillance registries and represents added value both for patients who are facilitated to participate and for companies who want an extension of indications, for example for rare diseases.
HOW TO CREATE A SUSTAINABLE CLINICAL PROGRAM

Special attention in registries is given to patient 2, because the first priority is to demonstrate benefits with a direct impact on health and quality of life. This can be done with the help of special disease-specific quality-of-life and symptom questionnaires, which must be validated in each individual language. The value and importance of such tools are enormous if they can provide reliable and reproducible data.
Large post-market studies require modern and comprehensive clinical data collection solutions. Technically, records can be mapped excellently in “electronic data capture” (EDC) systems. Digital tools today offer the ability to simplify clinical data and diagnostic image capture, remote monitoring, automated data analysis, and security assessment. In addition, Apps are now available to download to one’s cell phone, where the patient can give consent and does not even have to go to the testing center to collect questionnaires. Of course, data protection laws and regulations must be strictly adhered to, all for safety in the interest of the patient.
Author: Dr. Kristin Forßmann, director of clinical development Meditrial GmbH, Berlin, Germany
Modular protocol template (Table of contents). Ziegler et al, 2021






Leave A Comment